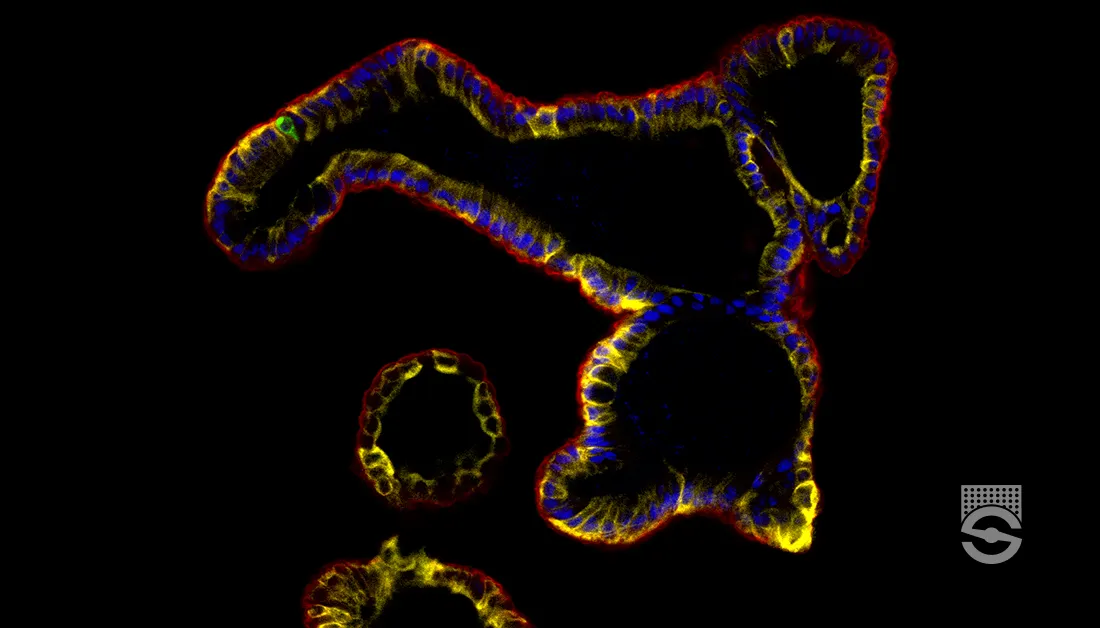
Overview
What is Fluorescent Live Cell Imaging?
To investigate cellular and subcellular processes, we can fluorescently label and visualize cells (or their components) of interest. Fluorescent molecules—dyes, probes, indicators, sensors, and natively expressed fluorochromes—that label the target are excited by an external light source then detected by a fluorescent microscope. By observing changes in fluorescence in the intact cell in real time, we can infer a lot about the dynamic cellular processes occurring in the microscopic world around us.
Importance of Imaging Live Cells
Fluorescent live cell imaging changed the game for biology research, allowing scientists to go from imaging only fixed cells (like viewing a static photograph) to incredibly dynamic cellular processes (like watching a movie). Advances in engineering fluorescent molecules as well as in microscopy enable us to use fluorescent live cell imaging to study cellular and subcellular changes in real time, furthering our understanding of physiological mechanisms in health and disease.
Fluorescent Molecules for Live Cell Staining
Advantages, Disadvantages, and Considerations
Fluorescent molecules confer high specificity for the accurate labeling, imaging, and analysis of proteins and cellular structures of interest. This degree of specificity and the ability to image changes in real time—in other words, high spatial and temporal resolution in living cells—are the main advantages of fluorescent live cell imaging.
When choosing a fluorescent molecule for live cell imaging, we consider the following aspects (Ettinger):
- Emission spectra should match the microscope’s filters
- Molecule should not aggregate or be toxic to the cells
- Maturation time should be appropriate for the process being studied, e.g. in an experiment assessing protein turnover, the fluorescent molecule should have the kinetics to bind and unbind quickly
- Molecule should not interfere with the function of the target, e.g. the FP moiety should be attached to a protein far from the site of enzymatic activity so that endogenous function is not altered
- Molecule should undergo as little photobleaching as possible
These considerations also speak to the potential disadvantages of fluorescent live cell imaging that need to be accounted for during experimental design, including toxicity of the fluorescent molecule to the cell, overexpression artifacts like aggregation, and nonspecific labeling by the molecule due to its aggregation or its nonspecific binding to intracellular components (Ettinger; Wang). Additionally, during imaging, fluorescent molecules can exhibit photobleaching (loss of fluorescence), show high background fluorescence, or render a low signal-to-noise ratio of the target, making successful imaging challenging. These disadvantages of the method can be overcome by testing and optimizing fluorescent molecules under the experimental conditions and with the microscope we intend to use for our study. For example, we can optimize the concentration of fluorescent dye and incubation time during staining (to prevent nonspecific labeling), and the sample illumination and imaging speed during image acquisition (to prevent photobleaching but still enable target detection).
Types of Molecules: Fluorescent Proteins, Dyes, and Tags For Live Cell Imaging
There are three main types of molecules to select from for fluorescent live cell imaging. Genetically encoded fluorescent proteins (FP) are a popular option. They need to be engineered, cloned into the desired cell model, system, or line, and optimized for the specific target. The second option is chemical fluorophore conjugation with fluorescein, rhodamine, or Alexa dyes. Modern dyes have higher signal-to-noise ratio compared to FPs, but they are also less feasible because they require purification, fluorescent labeling, and microinjection of the labeled protein (Ettinger).
The third option is a hybrid system that combines genetic encoding with chemical conjugation, allowing expression of a fusion protein made up of the target protein and a “tag” like SNAP-, CLIP- or Halo (Wang). The tag is the additional polypeptide or protein, which undergoes a specific reaction in the presence of a synthetic fluorescent molecule, leading to an observable change in fluorescence that we can use to infer target activity. The SNAP-tag, for example, undergoes an irreversible reaction with benzylguanine derivatives where it transfers the fluorophore-labeled benzyl group to an active-site cysteine residue. While such small-molecule fluorescent probes bypass the need to use invasive techniques like microinjection or electroporation, a disadvantage of this approach is that, with diameters of about 3-4 nm, tags can still affect the protein’s endogenous function and lead to mislocalization.
Applications of Fluorescent Live Cell Imaging
Different fluorescent molecules have been engineered and optimized for live cell imaging. To image proteins, we can use FPs that are photoswitchable (photoactivatable or photoconvertible) for pulse-chase and short-term experiments over a few minutes or FPs that are time-encoded (rely on maturation kinetics) for long-term experiments over a few hours (Knop). For imaging nucleic acids, we can leverage molecular beacons, dye aptamers, nanoflares, CRISPR/fluorophore-tagged Cas9, and other fluorescent probes. This equips us to (1) study transcription, translation, and DNA replication and repair, (2) perform drug-resistance monitoring during chemotherapy, or (3) detect predisposition to cancer (Specht). We can also use sensors for small molecules, metabolites, ions, and enzymatic activity.
In addition to imaging proteins and enzymes, DNA and RNA, small molecules, metabolites, and ions, there are fluorescent molecules that can tell us more broadly about the cell state, such as by detecting voltage and membrane potential, cell cycle phase, or the redox environment within the cell. Furthermore, fluorescent molecules can be “custom designed” for specific applications beyond those listed here: they can reveal the cellular identity as well as the morphology, dynamics, and activity of the overall cell or of its subcellular compartments. An example of using live cell imaging for cell identity exploits the NeuroFluor™ NeuO dye, a membrane-permeable fluorescent probe that selectively and reversibly labels stem cell-derived neurons in the presence of other brain cells (Er).
Imaging the Cell Cycle
To track the cell cycle for insights into states like cancer or aging, other than imaging DNA, we can take advantage of fluorescent molecules that initially localize to the nucleus but change their fluorescence to indicate the corresponding cell cycle phase. The Fucci sensor exhibits red fluorescence in G1 but shifts to green fluorescence in S, G2, and M phases (Marx; Specht). Other molecules change their location, translocalizating from nucleus to cytoplasm in accordance with the cell cycle, e.g. a CDK2-dependent sensor localizes to the nucleus in G0 and G1, to both the nucleus and cytoplasm in S phase, and exclusively to the cytosol in G2 and M when CDK2 activity is the highest (Specht).
Imaging Cell Viability
To characterize cell viability, one of the most straightforward approaches is using fluorescent cell viability dyes. Some examples include GloCell™ Fixable Viability Dye (e.g. Violet 540, Dye Red, etc.) or Trypan Blue (Liesche; Kerschbaum). GloCell, for example, works by irreversibly binding intracellular and cell surface amine groups (STEMCELL). Since cells with compromised plasma membranes become more permeable to the dye, dead cells will show greater fluorescence in a cell viability assay.
Imaging Subcellular Compartments
To examine subcellular processes, we can employ fluorescent molecules that have an affinity for particular organelles due to the protein moieties or unique chemical composition of the organelles. For instance, LysoBrite™ dye is a hydrophobic, fluorescent dye that permeates live cells, selectively accumulates in the lysosomes due to its pH gradient, and increases its fluorescence within the lysosome’s acidic environment (STEMCELL). Therefore, LysoBrite™ allows us to image the most active lysosomes. Similarly, the mitochondrial superoxide dye selectively targets the mitochondria of living cells. Since it oxidizes when it reacts with a superoxide, emitting red fluorescence, we can use the dye to observe the state of the cell, i.e. its redox environment, as well as to visualize an organelle (the mitochondria) (STEMCELL).
Considerations For Developing Specific Live Cell Imaging Protocols
Developing and optimizing a protocol for our specific live cell imaging application can be daunting. We must begin with a thorough understanding of our research question and conditions, including what we hope to track, which cells we are working with, and the specifics of our imaging setup e.g. which microscope filters are available to us? Based on this, we can comb the existing literature to find an appropriate fluorescent molecule—one that will be non-toxic for our cells, spatially and temporally specific for our target, and have a high signal-to-noise ratio for our particular application. Since there are so many different fluorescent molecules, the details for the protocols we use can vary immensely. We can rely on existing documentation like product information sheets and published work to figure out our overall protocol, such as medium and reagents to use, incubation times, microscope settings, and the length of the experiment.
Then, like all fluorescent imaging approaches, taking the time to get to know your microscope, having positive and negative controls, and setting up a few different experimental conditions will enable us to optimize the protocol for our application. If we see a good signal-to-noise ratio for our target during imaging, then success! But if there is not enough signal, we can turn to our controls to narrow down the source of the issue to autofluorescence, background fluorescence, physiological conditions of imaging (e.g. changes in temperature or pH), or lack of target specificity by the fluorescent probe. Worst case scenario, if we are unable to optimize a protocol for our live cell imaging application, the answer may lie in engineering a brand new fluorescent molecule!
References
- Ettinger A & Wittmann T. (2014) Fluorescence live cell imaging. Methods Cell Biol. 123: 77–94.
- Er JC et al. (2015) Neuo: A fluorescent chemical probe for live neuron labeling. Angewandte Chemie – International Edition 16;54(8):2442–2446.
- Liesche J et al. (2015) Cell wall staining with Trypan blue enables quantitative analysis of morphological changes in yeast cells. Front. Microbiol. 6(107) doi: 10.3389/fmicb.2015.00107.
- Knop M, Edgar BA. (2014) Tracking protein turnover and degradation by microscopy: photo-switchable versus time-encoded fluorescent proteins. Open Biol. 4(4):140002.
- Kerschbaum HH et al. (2021) Trypan Blue – Adapting a dye used for labelling dead cells to visualize pinocytosis in viable cells. Cell Physiol Biochem 55:171-184.
- Marx V. (2017) Cell biology: tracking a cell’s cycle. Nature Methods 14: 233–236.
- Specht EA, Braselmann E et al. (2017) A critical and comparative review of fluorescent tools for live-cell imaging. Ann Rev Phys 79: 93–117.
- STEMCELL.com (Accessed 2023) GloCell™ Fixable Viability Dye Violet 540.
- STEMCELL.com (Accessed 2023) LysoBrite™ Lysosome Dyes.
- STEMCELL.com (Accessed 2023) Mitochondrial Superoxide Dye.
- Wang L, Frei MS et al. (2018) Small-molecule fluorescent probes for live-cell super-resolution microscopy. J Am Chem Soc 141: 2770–2781.