Immunostaining Cryosections of Neural Organoids
This protocol is for immunostaining cryosectioned neural organoids to detect specific proteins or characterize cell phenotypes.
Before immunostaining, fix and cryosection neural organoids:
Expected Results
Expected results depend on the targets of interest, as immunostaining must be optimized for antigens, antibodies, cell and tissue type. Best practice is to refer to the Product Information Sheet as a starting point, and to optimize conditions in-house.
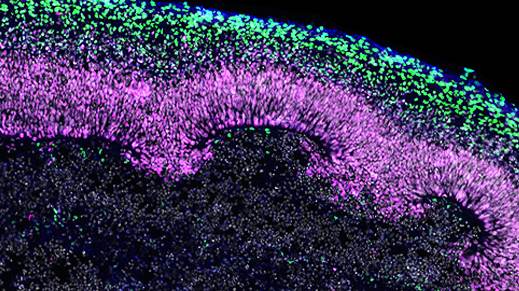
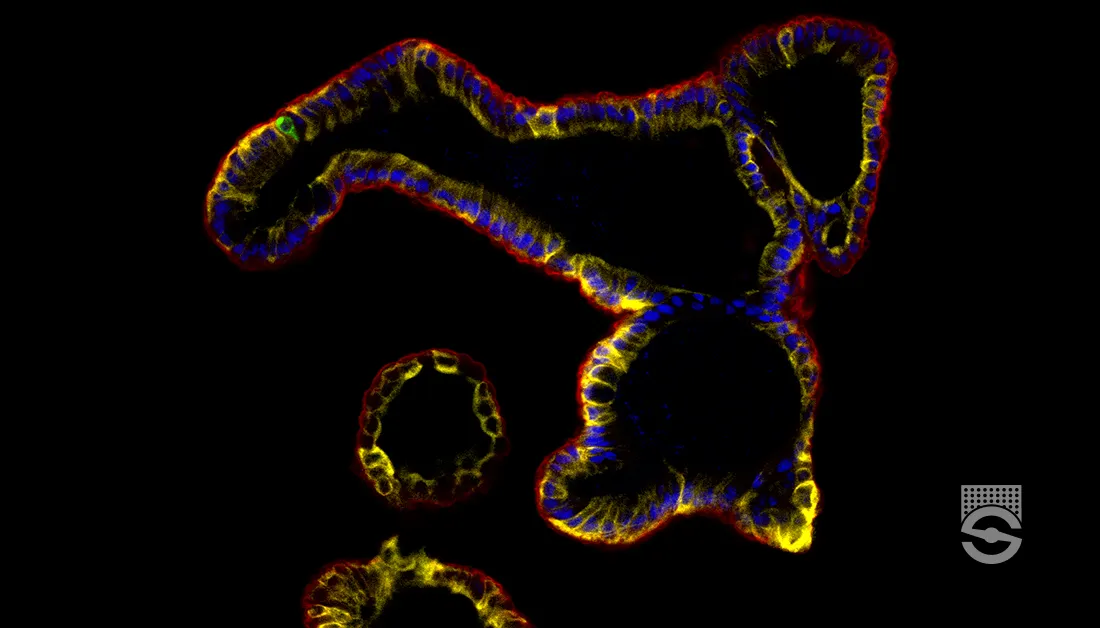
Figure 1. Immunofluorescence for CTIP2, PAX6, βIII-tubulin/TUJ1, and DAPI Showing Stratification of Cortical Plate Neurons and Progenitor Zones in Day 40 Cerebral Organoids
Day 40 cerebral organoids generated using the STEMdiff™ Cerebral Organoid Kit were cryosectioned into a 16-μm-thick section and immunostained for chicken ovalbumin upstream promoter transcription factor-interacting protein 2 (CTIP2; in green), paired box 6 (PAX6; in magenta), βIII-tubulin/TUJ1 (in blue), and DAPI (in gray). Cortical regions are defined by PAX6+ progenitor cells that are radially organized around a pseudo-ventricle. These progenitors give rise to cortical plate neurons indicated by CTIP2 and TUJ1 expression. This image was contributed by STEMCELL Technologies.
Materials
- Hydrophobic barrier PAP pen
- Coplin staining jars
- PBS-T, mixed thoroughly and can be stored at room temperature (15 – 25°C)
Material Amount Tween® 20 1 mL D-PBS 1 L - Blocking solution, prepared fresh, mixed thoroughly, and used immediately
- Normal Donkey Serum (NDS) diluted in PBS-T to a final concentration of 5%
- Primary dilution buffer, mixed thoroughly and can be stored at room temperature (15 – 25°C)
Material Amount Sodium azide in 100 mL PBS-T 0.05 % (final concentration) Bovine Serum Albumin (BSA) 100 mL - Primary and secondary antibodies diluted in the immunofluorescence buffer at appropriate dilution factor
- Can be identified with the Antibody Dilution Finder
- Coverslip
For Optional Antigen Retrieval
- Food steamer
- Heat-resistant plastic Coplin staining jar
- Tri-sodium citrate (dihydrate)
- 1 mL citrate buffer, stored at 2 – 8°C for up to 6 months
Material Quantity dH2O 950 mL Tri-sodium citrate (dihydrate) 2.94 g HCl / NaOH to adjust pH 6.0 pH Tween® 20 0.5 mL
Tissue Sample
Prepare Slides
- Remove sectioned slides from the freezer and allow to dry at room temperature.
- Outline sections with a PAP pen. When the pen wax is completely dry, proceed to the next step.
- If necessary, perform antigen retrieval. Otherwise, proceed directly to blocking.
Protocol
Antigen Retrieval (Optional)
Antigen retrieval is optional, but highly recommended. Cross-linkages formed by PFA fixation may interfere or block antibody binding to the epitope of interest so detection of certain samples will require an additional antigen retrieval step before incubation with primary antibodies.
- Place slides in a heat-resistant plastic Coplin staining jar and fill with citrate buffer.
- Place Coplin jar in food steamer and steam for 20 minutes.
- Carefully remove hot citrate buffer and wash slides 3X in PBS-T for 10 minutes each.
Blocking
- Wash slides with PBS-T at 37°C for 10 minutes to fully remove gelatin from slides.
- Lay slides flat and pipette enough blocking solution to completely cover sections. The borders formed by the wax pen will hold blocking solution on the sections of tissue.
- Incubate at room temperature in a humidified chamber for 1 hour.
Primary Staining
- Prepare primary antibody solution by adding primary antibodies at the appropriate dilution factor to the primary dilution buffer.
- Pour blocking solution off of slides and replace with primary antibody solution.
- Incubate at room temperature in a humidified chamber overnight (16 hours).
Secondary Staining
- Prepare secondary antibody mixes in PBS-T.
- Pour primary antibody mix off of slides and place slides in a Coplin staining jar.
- Wash slides 3X with PBS-T.
- Lay slides flat and pipette secondary antibody mix onto the slides. Incubate at room temperature in a humidified chamber for 2 hours.
Coverslips
- Pour off secondary antibody mix and place slides in a Coplin staining jar.
- Wash slides 3X with PBS-T for 30 minutes each.
- Air-dry slides for 5 minutes.
- Add PermaFluor™ to slides and add coverslip. Store at 2 – 8°C and allow to dry before imaging.
Notes
Safety Note
Prior to handling any chemical, consult the Safety Data Sheet (SDS) for proper handling and safety precautions.
References
- STEMCELL.com (Accessed 2024)
Cryogenic Tissue Processing and Section Immunofluorescence of Neural Organoids